Covid-19: SARS-CoV-2 variant B.1.1.7
Briefing: What is known about variant B.1.1.7?
Christmas was all but cancelled for much of the UK because a new variant of the Coronavirus had been identified which seemed to explain the significant rise in infections observed throughout London and the South East during early December 2020. There was much comment in the media, but not much by way of scientific explanation in terms of the changes this new variant has, what they might mean, and how it might be different to previous variants. Even the name of this new variant wasn’t made clear. This briefing reviews the scientific literature in an attempt to answer such questions.
It should take you about 30 minutes to read this post. You should be able to follow the material with only a basic knowledge of biology at the cellular level, but may find my post Genes, proteins & biological function a useful introduction.
- Introduction to SARS-CoV-2
- Brief introduction to SARS-CoV-2 variants
- How variants emerge
- Naming of mutations and variants
- Lineage of variants
- Variants of SARS-CoV-2 Emerging in 2020
- SARS-CoV-2 variant of concern: B.1.1.7
- Mutations in variant B.1.1.7
- B.1.1.7 mutations with particular significance
- Detection of B.1.1.7
- Clinical Impact of B.1.1.7
- Spread of B.1.1.7 beyond UK
- General Implications
- Conclusion
- Related Posts
- References
Introduction to SARS-CoV-2
Origin of SARS-CoV-2
The disease Covid-19 was first identified in the city of Wuhan (China) in late 2019. Researchers identified that it was caused by a new strain of the beta coronavirus (CoV) previous strains of which had been identified as causing diseases in bats as well as being responsible for epidemics of SARS in China and of MERS in the Middle East [Ref 1]. This new strain was officially named as SARS-CoV-2 by The World Health Organisation (WHO) on 11 Feb 2020 [Ref 2].
New strains of a virus are usually given an official name by WHO when the change in its genome is significant enough to cause some distinct functional difference in the associated disease. Changes to a virus strain are called variants and a variety of different naming systems are used to identify them.
Genomic structure of SARS-CoV-2
SARS-CoV-2 is a non-segmented RNA virus and when first identified in January 2020 had 29,903 nucleotides [Ref 3]. Its genome, as summarised in Figure 1, contains a number of regions that code for viral structural proteins common to the coronavirus (CoV) family of viruses: spike (S), envelope (E), membrane (M), nucleocapsid (N). There are also coding areas for 16 non-structural proteins some of which are common to other CoV viruses. These non-structural proteins are involved in its replication and are also associated with causing disease in the host organism [Ref 4,5]. Finally there are nine regions that code for accessory proteins whose role is less well understood [Ref 6].
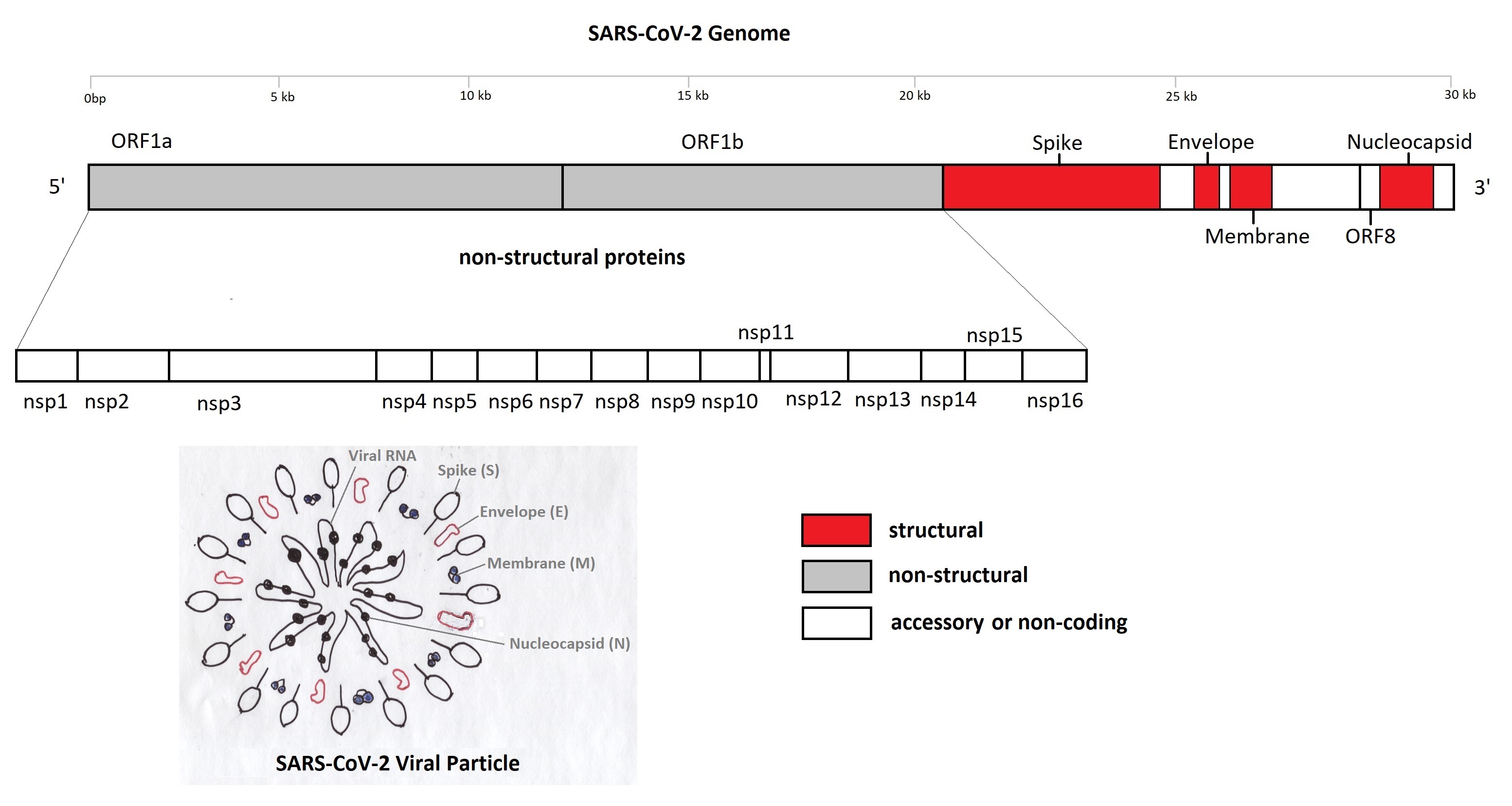
Figure 1: Significant Proteins of the SARS-CoV-2 Genome [Ref 7]
Viruses like SARS-CoV-2 in themselves lack some of the properties deemed essential for classification as a separate organism. Instead they use the machinery found in their human host cells to do things like replicate or generate the energy they need. Accordingly the term virus particle it is more appropriate rather than virus cell.
SARS-Cov-2 Mutation rate
RNA viruses must hijack mechanisms in their hosts cells in order to replicate themselves. When this happens the chance of an error whilst copying any base molecule in their genome can be as high as 1 in 10,000 [Ref 8]. These errors, or mutations, then become copied in turn so the virus genome acquires further mutations at each generation. Such a high mutation rate allows the virus to adapt rapidly and exploit changes in its environment, but it also makes the virus prone to lethal mutation; mutations that stops it functioning or replicating. It is for this reason that many mutations do not manage to replicate further. In fact the observed rate of conserved mutations in SARS-CoV-2 is estimated at only two nucleotide mutations per month [Ref 9]. This is lower than expected for an RNA virus with such a large genome. It may be explained by the presence of the non-structural protein nsp14-ExoN found in the CoV family of viruses which seems to provide a proof reading mechanism that acts to increase the fidelity of base molecule copying during replication [Ref 9].
result in some areas of the genome becoming identified as mutational hotspots
Although nucleotide mutations arise by chance throughout the SARS-CoV-2 genome, some bases like cytosine are more prone to mutation than others and certain sequences are similarly more to suffer mutations than others [Ref 10]. There are also certain areas of the genome in which mutations tend to be observed less often than others due to selective pressures. For example mutations in some locations of the coding area for a given protein might more readily lead to lethal mutation, whilst mutations in other locations might be better tolerated or repaired. Such factors, and others, result in some areas of the genome becoming identified as mutational hotspots [Ref 11].
Brief introduction to SARS-CoV-2 variants
How variants emerge
The process of natural selection ensures that SARS-CoV-2 virus particles containing a set of mutations which somehow improve their ability to replicate and thrive will become more numerous in a given local population. Eventually virus particles sharing a set of advantageous mutations will start to dominate the local population. This marks the emergence of a new variant of the virus. In time this new variant will itself become subject to the forces of natural selection as another variant starts to emerge; one which is even better adapted to the environment. This variant might arise from some mutations suffered by its predecessor, or might be non-related variant that has been imported from a different local population.
Classifying virus particles according to the set of mutations they contain is a useful way to study the emergence of new variants. The whole genome sequence of SARS-CoV-2 can be determined from clinical respiratory samples using the same Next-Generation Sequencing (NGS) methods commonly used in genomic research. The resulting sequences are usually deposited in publicly available databases like GISAID and EpiCov so new variants can be identified and their progression traced as a global effort.
A total of 93,817 SARS-CoV-2 sequences had been deposited in the GISAID database by 6 September 2020. The UK accounted for the majority of these (38.9%), followed by USA (22.7%) with Australia, Spain, and India also making significant contributions [Ref 12]. The large numbers of samples being sequenced in the UK means that new variants of the virus in this country are more likely to be detected sooner than elsewhere.
Naming of mutations and variants
Mutations are often named by the location and type of a nucleotide mutation. For example A23403G is a mutation arising from the substitution of the base adenine (A) for guanine (G) which is 23,403 bases from the start of the SARS-CoV-2 genome. However, when a mutation starts to become more frequently observed in a population it is identified as an emerging variant, traditionally according to the resultant change in the amino acid of the protein for which it codes. For example, the nucleotide mutation A23403G results in S.D614G, a change of aspartic acid (D) to glycine (G) in the 614th codon of the amino acid chain that codes for the spike protein (S). Accordingly the variant is named D614G.
In the case of most viruses the mutations that give rise to variants involve changes to only one or two amino acids so a naming system based on the amino acid change is reasonable, but it becomes less so when there are multiple amino acid changes and new variants are emerging each month; the case in SARS-CoV-2. Therefore names are used which reflect the emerging variant’s origins (lineage). For example, the Pangolin system names D614G as B.1 showing that it is the first emerging variant from the B lineage linked to Wuhan [Ref 13] whereas in the Nextstrain system it is named 20I/D614G reflecting that variants in this branch are characterised by the S.D614G mutation whose parent was the ninth (I) SARS-CoV-2 variant to become dominant in 2020 (20) [Ref 14]. However, these naming systems struggled to keep pace with the changes in SAR-CoV-2 that are were happening throughout the world in 2020.
Nextstrain was obliged to create a number of new branch (clade) names in early January 2021, including 20I which resulted in the new name 20I/D614G for what was previously called 20B/D614G. Some Nextstrain clade names have also acquired a number to indicate a variant of concern, for example 501Y.V1 (UK) and 501Y.V2 (South Africa) and 501Y.V3 (Brazil).
The Nextstrain and Pangolin naming systems both have their draw-backs, so not surprisingly neither has been adopted widely adopted outside the scientific community. Indeed, even some scientists have criticised the way SARS-CoV-2 variants are named; a situation described as a ‘bloody mess’ by the influential journal Nature [Ref 15]. Perhaps as a consequence, the UK NERVTAG group experts have adopted a variant name system for SARS-CoV-2 that avoids including details of lineage. Instead it gives a general classification, followed by the year and month, followed by a unique number. Therefore in this system the variant 501Y.V1 is named VOC 202012/01 indicating it is the first ‘Variant Of Concern’ named in December 2020.
Lineage of variants
Studying the genetic differences between samples of viruses provides useful information not only about the relative dominance of a particular variant, but also about its lineage in terms of when and where the mutation first arose as well as its relationship to earlier mutations.
A study by Koyama, Platt, and Parida [Ref 16] in May 2020 analysed the genetic sequences of 10,022 SARS-CoV-2 virus samples deposited from 68 countries in various databases against the reference genome NC_045512 [Ref 3]. The study found 65,776 mutations of the virus, but only 5,775 were identified as distinct variants. The second most frequently observed non-synonymous variant was S.D614G, observed in a total of 6294 samples. The locations and dates from which the samples containing the S.D614G variant were analysed together with the other variants. This work allowed the construction of a phylogenetic tree tracing the relationship between all the major mutations back to the reference genome, sequenced in December 2019 from samples obtained in Wuhan [Ref 3]. It initially had six major branches, or clades, the largest being S.D614G which had five subclade. However, many new clades were subsequently added as the virus spread and mutated.
The Nextstrain website produces a similar phylogenetic tree which displays the lineage of variants identified from SARS-CoV-2 genome sequences that have been uploaded to the GISAID database. A screenshot of this website is shown in Figure 2. It’s tree displays the lineage of the major clades classified by Nextstrain in December 2020 together with the emerging clades arising from them. The corresponding map shows that the 20B clade is dominant in UK, whilst the 20A clade is more dominant in other parts of Europe.
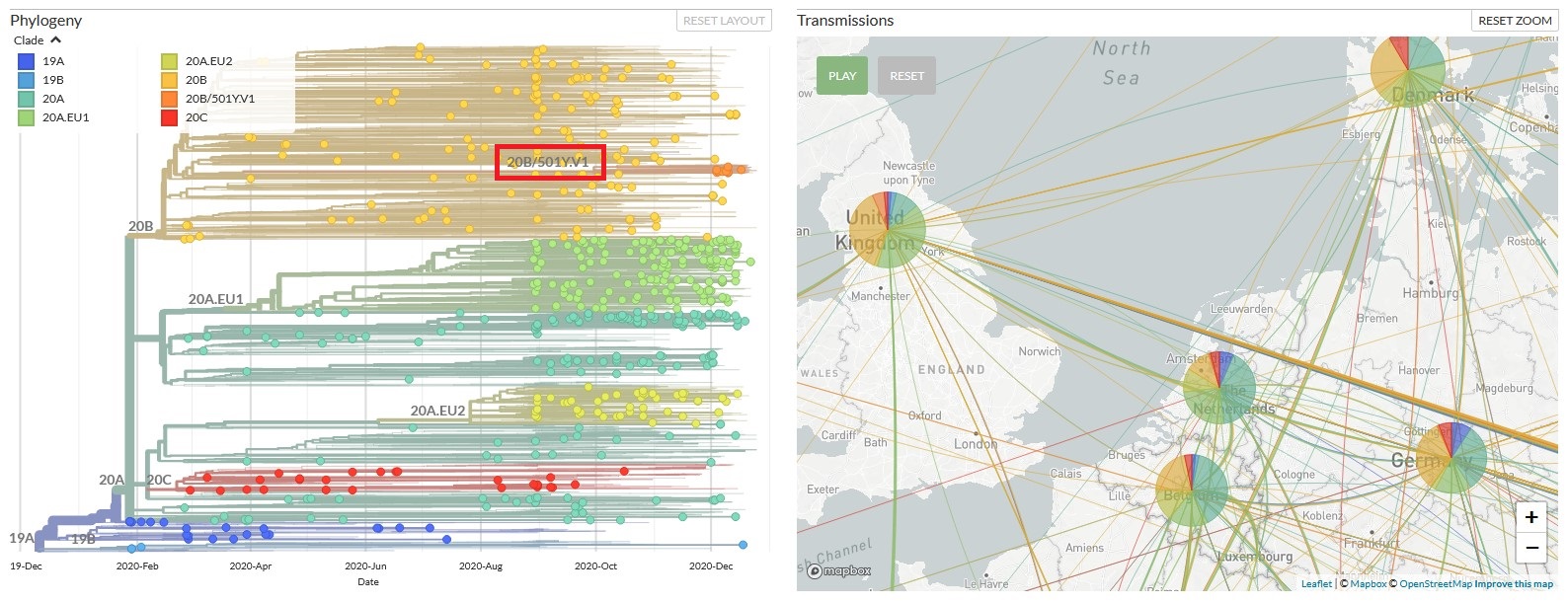
Figure 2: Lineage of SARS-Cov-2 B.1.1.7 classified by Nextstrain in December 2020
Variants of SARS-CoV-2 Emerging in 2020
SARS-Cov-2 is the first Coronavirus to cause a global pandemic. Although its mutation rate is low compared to other RNA viruses, it prevalence in such a large number of local populations has resulted in the emergence of many variants in the 12 months since it was first identified. Some of the most notable include:
20A (B.1)
It is defined by the amino acid mutation S.D614G. It emerged in January 2020 and soon became the dominant lineage in Europe before spreading to the United States, Canada and Australia.
Lab experiments suggest the mutation enhances viral load in the upper respiratory tract so may increase the chance of infection, though it was not found to reduce the effectiveness of vaccines [Ref 17].
20B (B.1.1)
It is defined by the additional amino acid mutations N.R203K, N.G204R, ORF14.G50N. It emerged in February 2020 and some child variants have become the dominant strain in UK. One of particular interest is the main subject of this post; B.1.1.7.
20E.EU1 (B.1.177)
It is defined by the amino acid mutation S.A222V was reported in Oct 2020. It is characterised by additional mutations; ORF10:V30L, N:A220V, ORF14:L67F. Sequences containing this variation date from 20th June in Spain and by autumn had become the dominant variant in many European countries as well as being present in Hong Kong, Australia, New Zealand and Singapore. It is thought that this variant was spread rapidly by summertime travellers.
The A222V mutation occurs in an areas of the Spike protein not concerned with binding to human cell receptors. Lab studies failed to show any enhanced transmissibility in humans, though they were simplified and did not involve the entire SARS-CoV-2 virus [Ref 18].
20H/501Y.V2 (B.1.351)
It is characterised by amino acid mutations in the Spike protein: N501Y, K417N, E484K and 6 other non-synonymous mutations; 9 changes in total. It was reported 18 December 2020 in South Africa, but seems to have become dominant in October 2020. The emergence of this variant from clade 20H suggests it has arisen independently of 20I/501Y.V1 (B.1.1.7) which emerged from clade 20I, but shares the same S.N501Y amino acid mutation.
The variant 20H/501Y.V2 has been associated though genomic and epidemiological studies with a significant increase in transmissibility, possibly due to the S.N501Y and S.E484K mutations in the receptor binding domain (RBD). Furthermore both these mutations may contribute towards immune evasion by the RBD so have potential to allow re-infection or reduce the effectiveness of vaccines [Ref 19].
The non-synonymous mutations in the B.1.351 variant are: ORF1ab: K1655N; Envelope: P71L; Spike: N501Y, K417N, E484K, D80A, D215G, A701V; Nucleocapsid: T205I [Ref 62]
20J/501Y.V3 (P.1)
It is characterised by amino acid mutations S.N501Y, S.K417N, S.E484K, 12 other non-synonymous mutations, ORF1ab: SGF 3675-3677 delete, and 4 synonymous mutations; 20 changes in total. It was reported January 2021 in Japan, but originates from Manaus, Brazil. The emergence of this variant from clade 20J (B.1.1.28) suggests that it has arisen independently of both 20I/501Y.V1 (UK) and 20H/501Y.V2 (South Africa) though it shares some of the same mutations; both have S.N501Y whilst 501Y.V1 shares the SGF 3675-3677 delete and 501Y.V2 shares S.K417N as well as S.E484K [Ref 20].
501Y.V3 was absent from the publicly available sequences from samples collected in Manaus from March to November 2020, but seems to have spread quickly in December suggesting all its mutations arose at the same time. Early epidemiological data showed that it has high transmissibility and it was also speculated that presence of the S.E484K mutation might allow re-infection [Ref 21]. Later epidemiological data also suggested that re-infection may have occurred, though other causes might also explain the high number of new infections in a local population thought to have acquired herd immunity [Ref 52].
The non-synonymous and delete mutations in the P.1. variant are: ORF1ab: S1188L, K1795Q, del SGF 3675-3677; Spike: S.N501Y, S.K417N, S.E484K, L18F, T20N, P26S, D138Y, R190S, H655Y, T1027I; ORF3a: G174C; ORF8: E92K; Nucleocapsid: P80R [Ref 63]
SARS-CoV-2 variant of concern: B.1.1.7
Mutations in variant B.1.1.7
A new SARS-CoV-2 variant was identified in UK, December 2000. The emerging variant is named VOC 202012/01 in some government documents, but is called B.1.1.7 in the Pangolin system. Nextstrain initially named it 20B/501Y.V1, but later changed the name to 20I/501Y.V1 [Ref 22]. The first sample of this variant was sequenced 20 September 2020 and came from a patient in Kent. It has been subsequently suggested that all its characteristic mutations emerged over a short period in the same chronically-infected and immune-deficient/suppressed patient [Ref 23]. When this first sample was sequenced there was no reason to suppose that its mutations would characterise a new variant or confer any advantage to the virus like increase in transmissibility.
B.1.1.7 is exceptional as it contains 23 nucleotide mutations in the protein coding areas of the SARS-CoV-2 genome. Six of these mutations do not change the resultant amino acid (synonymous). The remaining 17 mutations result in the following changes to the amino acids used to build the various proteins of the virus [ref 39,47]:
- ORF1ab mutations: T1001I, A1708D, i2230T, SGF 3675-3677 delete. ORF1ab contains 16 non-structural proteins as shown in Figure 1. These proteins are mainly enzymes thought to be concerned with catalysing the replication of the virus RNA in human host cells as well as suppressing the host immune response and host gene expression [Ref 24] . Therefore these mutations have the potential to change transmissibility.
- Spike (S) mutations: N501Y, P681H, HV69-70 delete, Y144 delete, A570D, T716I, S982A, D1118H. The S protein contains two functional subunits. The S1 subunit contains a fragment called the receptor binding domain (RBD) which binds to the angiotensin-converting enzyme 2 (ACE2) protein in a human host cell. The S2 subunit is responsible for fusion of virus and human cell membranes [Ref 25]. Therefore S mutations, like those in the ORF1ab, have the potential to change the transmissibility of the virus.
- ORF8 mutations: Q27 Stop, R52I, Y73C. The ORF8 protein is not thought to be concerned with the functioning of virus itself or its replication, but together with the N protein seems to create disruption of key metabolic pathways in human host cells [Ref 26]. Therefore mutations in ORF8 may increase or decrease the severity of the Covid-19 disease.
- Nucleocapsid (N) mutations D3L, S235F: The N protein surrounds the virus genome and is essential for replication of the virus in the human host cell. The N protein together with the ORF8 protein seems to create disruption in human host cells [Ref 27]. Therefore mutations in N may increase or decrease the transmissibility of the virus as well as the severity of the Covid-19 disease
all these hyper-mutant variants appeared within the final quarter of 2020...
Most branches of the SARS-CoV-2 phylogenetic tree show no more than a few mutations and such mutations have previously accumulated at a relatively constant rate of 1-2 mutations per month [Ref 9]. Therefore the accrual of 14 amino acid replacements and 3 deletions in the B 1.1.7 variant (20I.501Y.V1) was unprecedented [Ref 28], at least until the report of the variants B.1.351 (20H/501Y.V2 South Africa)and P.1 (20J/501Y.V3 Brazil). The fact that all these hyper-mutant variants have appeared within the final quarter of 2020, yet seem to have emerged independently suggests a departure from typical viral evolution.
B.1.1.7 mutations with particular significance
The following mutations of B.1.1.7 in the Spike protein were identified by Rambaut A et al [Ref 28] as having potential biological effects of particular significance:
- S.N501Y – its location in the Receptor Binding Domain (RBD) region suggests it may change the binding affinity with the ACE2 protein in human host cells, so increasing transmissibility [Ref 29,30]. In addition it seems to be involved in interactions between the virus and antibodies produced by Covid-19 patients so has potential to reduce the effectiveness of vaccines[Ref 31,32]. The same mutation exists in the South African B.1.351 variant and the Brazilian P.1 variants, though they have a different lineage.
- S.HV69-70 delete – thought to enhance viral infectivity and have potential to reduce effectiveness of vaccines with or without the N501Y mutation. It has also been found in other variants including the S.Y453F variant (Denmark cluster 5) [Ref 33].
- S.P681H – suspected of assisting the entry of the virus into human host respiratory epithelial cells so has potential to increase transmissibility [Ref 34]. The same mutation exists in a variant with separate lineage originating in Nigeria, but does not exhibit the increase in transmissibility associated with B.1.1.7.[Ref 35]
It has been proposed that the B.1.1.7 variant, like the B.1.351 variant, may have arisen from prolonged incubation of viable mutations within individuals having a compromised immune system and suffering from chronic Covid-19 and may result from antigenic evolution [Ref 19,36,37] – i.e. it results from the selective pressure for the virus to escape recognition by the immune system of previously infected individuals.
Detection of B.1.1.7
Whole genome sequencing is necessary to identify all the actual mutations in a sample, but it is performed for only 5-10% of all positive PCR tests in UK and even less in most other countries [Ref 12]. Consequently whilst many countries have a good indication of the number of new cases of SARS-CoV-2 infection from their PCR data, they often lack information about what variants are present in this data. However, the B.1.1.7 variant was an exception. This was partly due to the relatively high level of sequencing undertaken in UK, and partly due to a fluke in the location of one of its characteristic mutations.
by luck includes the position of the HV69-70 delete...
TaqPath COVID-19 PCR test [Ref 38] used in many UK labs is comprised of three probes that cover small sections of sequences for proteins in three different areas of the SARS-CoV-2 genome. One of these probes covers a sequence that codes for the Spike (S) protein and by luck it includes the position of the HV69-70 delete mutation. Therefore this probe passes in samples that contain the Spike protein without the mutation, but fails in any sample that contains the HV69-70 delete mutation. This is called ‘S Gene Drop’, or 'Spike Gene Target Failure' (SGTF) and presents an opportunity to track the spread of B.1.1.7 using PCR data [Ref 39,40], but assumes:
- HV69-70 delete indicates the presence of the B.1.1.7 variant rather than some other variant with the same mutation [Ref 39]
- The other two probes either both pass, or both fail, so the overall result for the test for SARS-CoV-2 is correct [Ref 41,42].
The above assumptions were considered reasonable by Public Health England because sequence data had detected B.1.1.7 in a high number of samples over the period leading to 5 December which strongly suggested SGTF could not be attributed to another variant [Ref 40]. Consequently estimates of growth rate for the new variant were produced by combining data from PCR tests with sequence data. This analysis suggested B.1.1.7 exhibited a significant increase in transmissibility [Ref 39,40].
The HV69-70 delete required for SGTF is characteristic of the B.1.1.7 variant, but is not characteristic of the P.1 (Brazil) or B.1.351 (South Africa) variants. Therefore SGTF from PCR data cannot be used to track the spread of these other variants. However, a PCR test for a specific variant can be developed by creating special PCR probes particular to its individual mutations. This type of variant specific PCR test was used in early February 2021 to track the spread of the B.1.351 variant in the UK.
Clinical Impact of B.1.1.7
Following a meeting of the UK NERVTAG group of experts 18th December the variant was upgraded to a variant of concern (VOC 202012/01) [Ref 40]. Within days the UK Government took action to constrain the spread of the new variant, though sadly by early January transmission rates had soared. It is reasonable to suggest that without these actions the situation in the UK would have been much worse. Subsequent studies by Davies et al suggested B.1.1.7 was 56% more transmissible [Ref 43] whilst Volz et al observed a 75% advantage in transmission as well as a statistically significant shift in the infection of the under 20 age group [Ref 44].
for men aged 60 the mortality was 13 or 14 per 1,000...
Early analysis of epidemiological data suggested the hospital admission ratio as well as age, sex, reinfection and mortality profiles were not significantly changed by the new B.1.1.7 variant [Ref 39]. However, the UK NERVTAG group announced 21st January that increased mortality had been observed in people who had a positive PCR test with SGTF against those with non-SGTF. This was interpreted to show that variant B.1.1.7 is associated with an increased risk of death compared to other variants circulating the UK [Ref 45].
The UK chief scientific adviser, Patrick Vallance, used the data provided by the NERVTAG group to support claims that for men aged 60 the mortality associated with B.1.1.7 was 13 or 14 per 1,000 against 10 per 1,000 in other variants. Furthermore he claimed the variant was 30%-70% more transmissible than previous UK variants [Ref 46].
Spread of B.1.1.7 beyond UK
By January 28 2021 the B.1.1.7 variant was found in sequences uploaded to GISAID from 31,907 samples in 50 countries, though the majority were from UK (29,556) [Ref 47]. Therefore it is possible that many countries are now in the same situation as UK in early December 2020 with B.1.1.7 having established itself, but not yet causing a significant surge in infection.
The low levels of sequencing achieved in many countries makes it difficult to trace the spread of the variant using SGTF data with any certainty. For example in USA where SGTF was detected in 14 States between October and December 2020 [Ref 48], the first B.1.1.7 variant was only confirmed by sequencing 29 December 2020 [Ref 49]. Similarly Portugal has observed an increase in SGTF from 5.8% in December 2020 to 13.3% in mid-January 2021, but the total number of sequences was insufficient to conclusively attribute B.1.1.7 as the cause [Ref 50]. Therefore the low level of sequencing in both US and Portugal does not preclude the emergence of another variant with the same HV69-70 delete mutation.
PCR positive cases in Portugal surged from a 7 day average of 2,921 in 28 December 2020 to 12,417 by 26 January 2021 [Ref 51].
General Implications
Groups at risk
It is unclear why Covid-19 is more lethal to some people than others. A number of associations have been observed between the severity of disease and traits such as sex, age and existing medical conditions [Ref 52, 54], though similar associations exist in many diseases. There is also evidence from studies of patients in UK and US that people from Black and Asian ethnic backgrounds have worse clinical outcomes from Covid-19 than White individuals [Ref 55] though this is thought to arise from factors like differences in the socio-economic background rather than genetics. However, some evidence of genetic risks comes from a study that reports an association between severe Covid-19 and certain human genes carried by 50% of people in South Asia and 16% of people in Europe [Ref 56]. This suggests it may be possible to tailor treatment to individuals based on their genetic profile and the variant of the virus they have contracted.
Future variants
The B.1.1.7 variant of SARS-CoV-2 has emerged from earlier variants and may itself parent further variants which in turn may come to dominate local populations due to some enhanced trait like further increased transmissibility. Therefore it is possible that B.1.1.7 will become the source of many future variants and be identified as a major clade in 2021. However, a variant introduced from another local population with a particularly advantageous trait may out-compete B.1.1.7. In this case B.1.1.7 will eventually share the fate of many other variants like S.Y453F (Denmark) and stop circulating [Ref 57].
Our best hope is future variants become less virulent...
It is improbable that SARS-CoV-2 will simply disappear of its own accord, like the earlier Coronavirus associated with SARS in 2004. It is now just too difficult to isolate people with the virus in all local populations at the level required to stop its circulation. Similarly there are too many variants dominating different local populations to expect one single variant to emerge that would manage to out-compete all the others and then suffer a mutation which would stop it being able to replicate in human host cells. Our best hope is that future variants will become less virulent in terms of the disease they cause so we can tolerate living with SARS-CoV-2 in much the same way we live with the influenza virus, though improvements in immunity, whether acquired naturally or from vaccines, will undoubtedly have a key role in the future control of the virus.
Herd immunity
When more than a certain percentage of people in a local population acquire immunity to a virus, either by vaccination or previous infection, further infection drops dramatically as a consequence of an effect called herd immunity. However, allowing herd immunity to arise simply by letting people become infected would result in many deaths; for example mortality is estimated at 1 in 100 for men aged 60 [Ref 46]. There is also the question of how long immunity acquired from such infection would last as well as the possibility that a new variant may allow people to become re-infected anyway [Ref 52]. Therefore there is no real alternative to a global programme of vaccination.
Vaccination
Current vaccines like Oxford-AstraZeneca and Pfizer-BioNTech have been approved on the basis of their efficacy in terms of preventing severe symptoms of Covid-19, not preventing infection [Ref 58,59]. However, the large scale vaccination programmes that have been initiated in the UK and elsewhere should produce data about preventing infection by Q2 2021, if not before. It is to be hoped that these results will be as impressive as those obtained from clinical trials in respect of preventing serious illness.
There is concern about the effectiveness of vaccines on new variants, especially variants with mutations in the Spike protein which is the target of both the Oxford-AstraZeneca and Pfizer-BioNTech vaccine. There is particular concern about the S.E484K mutation that exists in the B.1.351 (South Africa) and P.1 (Brazil) because it has been associated with a small number of re-infection cases [Ref 52,60]. It is difficult to predict with any certainty the impact of any mutation, so the evidence of any reduction in the effectiveness of a vaccine will inevitably come from observational studies. These can only report when significant numbers of people have been vaccinated in a local population in which a variant with the mutation has achieved dominance. Therefore it is necessary to wait for such evidence.
Variations in the viral proteins targeted by vaccines are constrained by a need for them interact with proteins in their human hosts, so whilst such mutations may reduce the effectiveness of some vaccines, they are unlikely to render them entirely useless. However, even a small change in the Spike protein may give the variant an evolutionary edge in a vaccinated local population allowing it to dominate and then spread elsewhere. For this reason the overall success of vaccination depends on lowering the probability of a new variant emerging that reduces vaccine effectiveness. This means vaccinating quickly to reduce the time the virus has to mutate, having different types of vaccine to avoid just one mutation getting the necessary evolutionary edge, and taking measures to stop the virus spreading in order to lower the total number of virus infections from which a mutation can emerge. It also means addressing the cause of hyper-mutational SARS-CoV-2 variants like B.1.1.7 (UK), B.1.351 (South Africa), and P.1. (Manaus, Brazil).
Even if a local population has controlled the virus by vaccination, it remains vulnerable to a variant arriving from elsewhere especially one which has evolved some degree of vaccine resistance. Consequently it is essential that we quickly complete vaccination on a global basis and keep our local measures to stop the virus spreading until the virus has been successfully controlled everywhere.
Conclusion
What is known about B.1.1.7
Whilst there is precise knowledge about the SARS-CoV-2 genome and the mutations that characterise its mutations, science has not yet reached the point of being able to produce reliable predictions of functional outcomes based solely on such genomic data and models of proteomic behaviour. Consequently most information about the impact of the virus and its mutations on disease and its transmission must come from epidemiological and lab studies. Unfortunately such studies take time and often fail to product conclusive results. Therefore in the meantime we have to rely of the results of the initial studies, such as those published 21 January 2021, which suggest:
- the B.1.1.7 variant has higher transmissibility than previous variants and causes increased mortality, but otherwise is similar in terms of outcomes than previous variants [Ref 45].
It would not be surprising if future studies involving more extensive research and better data reach different conclusions to those given in these initial studies.
Open Questions
Despite the huge international research effort in 2020 a number of important scientific questions remain unanswered at the start of 2021:
- Do the approved vaccines reduce infection as well as severe illness? The clinic trials for the approved vaccines show clear efficacy in terms of reducing severe illness due to Covid-19, but will these results be confirmed by the data from large scale vaccination programmes? Just as importantly, will the data also show that vaccinated people are also less likely to transmit the virus to others?
- Might P.1 (Manaus, Brazil) or B.1.351 (South Africa) become dominant in UK? Will the traits of these other variants allow them to out-compete B.1.1.7? Alternatively will the UK measures to reduce infection and its vaccine programme stop them developing beyond a few isolated cases?
- Will current vaccines be effective against new variants? What will the data from large scale vaccinations show in terms of the effectiveness of vaccines against new variants? What might we learn about the potential of certain mutations to change vaccine effectiveness?
- How can new variants be better detected and tracked? the emergence of B.1.1.7 in a country that sequences a relatively high level of positive cases allowed it to be detected quickly. What sort of global effort is required to allow future variants to be detected and tracked so allowing timely interventions to be made?
- What caused the hyper-mutation in B.1.1.7? Does the high number of mutations in the B.1.1.7, B.1.351 and P.1 variants result from treatment given to chronically-infected and immune-deficient/suppressed patients? If so what changes in treatment or infection protection measures need to be taken?
- How long before B.1.1.7 spreads beyond UK? B.1.1.7 has already been detected in 49 countries besides UK, so will its higher levels of transmissibility allow this variant to out-compete other variants in their local populations and become dominant? How long before this happens?
- What is the ultimate fate of B.1.1.7? Will B.1.1.7 acquire further mutations that give it advantageous traits and so spawn further variants, or will it be supplanted by a variant from a different lineage and so stop circulating? How long will it take for such new variants to emerge?
- Why do young people not suffer from Covid-19? In previous pandemics involving a respiratory virus people with less effective immune system, both the very young and very old, had an elevated risk of mortality [Ref 61]. Therefore why do so many people under 18 years old suffer so little from Covid-19 ?
- Why do people react differently to Covid-19? Severe Covid-19 is associated with expected risk factors, like age and existing medical conditions. However it also seems that some people have genetic susceptibilities to the disease. Can these susceptibilities be identified by genetic markers so tests can be developed for them? Might a better understanding of these genetic factors allow a more effective and personalised treatment?
- What are the long term effects of Covid-19? The long term clinical outcomes of Covid-19 are unknown, though clearly the disease can affect multiple organs besides the lungs and may persist for many months. What will we discover about the disease in 2021 and beyond?
Is there a Covid Dividend?
Whilst SAR-CoV-2 has had a catastrophic impact on many people throughout world, it has also produced a few dividends. For example it has driven the development of new types of vaccine with potential for use against other types virus including those we have yet to encounter. It has also produced vast quantities of data which will help provide new knowledge and understanding about viruses and the disease they cause. However, perhaps the biggest dividend is it making a compelling case for global cooperation and a return to rational thinking.
If you found this post useful please help raise my profile by providing a link on your social media account; just click the buttons at the top of the page. You can also add a comment or contact me by email, but I can’t promise to respond immediately.
Related Posts
The following posts provide further information:
- Genes, proteins & biological function – a brief introduction to how life works
Further information
The following links provide information related to this post:
References
The following sources of information are referenced in the post:
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020 Feb 22;395(10224):565–74. Available from: https://pubmed.ncbi.nlm.nih.gov/32007145/
- WHO | Naming the coronavirus disease (COVID-19) and the virus that causes it [Internet]. Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it
- Wu F, Zhao S. Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome. 2020 Mar 18; Available from: http://www.ncbi.nlm.nih.gov/nuccore/MN908947.3
- Nakagawa S, Miyazawa T. Genome evolution of SARS-CoV-2 and its virological characteristics. Inflammation and Regeneration. 2020 Aug 10;40(1):17 Available from: https://inflammregen.biomedcentral.com/articles/10.1186/s41232-020-00126-7
- Yuan S, Peng L, Park JJ, Hu Y, Devarkar SC, Dong MB, et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Molecular Cell. 2020 Dec 17;80(6):1055-1066.e6. Available from: https://www.sciencedirect.com/science/article/pii/S1097276520307413
- Michel CJ, Mayer C, Poch O, Thompson JD. Characterization of accessory genes in coronavirus genomes. Virology Journal. 2020 Aug 27;17(1):131. Available from https://virologyj.biomedcentral.com/articles/10.1186/s12985-020-01402-1
- Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells [Internet]. 2020 May 20;9(5). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7291026/
- Sanjuán R, Nebot MR, Chirico N, Mansky LM, Belshaw R. Viral Mutation Rates. J Virol. 2010 Oct;84(19):9733–48. Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2937809/
- Smith EC, Blanc H, Vignuzzi M, Denison MR. Coronaviruses Lacking Exoribonuclease Activity Are Susceptible to Lethal Mutagenesis: Evidence for Proofreading and Potential Therapeutics. PLoS Pathog [Internet]. 2013 Aug 15 ;9(8). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3744431/
- Li Y, Yang X, Wang N, Wang H, Yin B, Yang X, et al. Mutation profile of over 4500 SARS-CoV-2 isolations reveals prevalent cytosine-to-uridine deamination on viral RNAs. Future Microbiology. 2020 Sep 1;15(14):1343–52. Available from https://www.futuremedicine.com/doi/full/10.2217/fmb-2020-0149
- Laamarti M, Alouane T, Kartti S, Chemao-Elfihri MW, Hakmi M, Essabbar A, et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. Gao F, editor. PLoS ONE. 2020 Nov 10;15(11):e0240345. Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0240345
- Furuse Y. Genomic Sequencing Effort for SARS-CoV-2 by Country during the Pandemic. International Journal of Infectious Diseases [Internet]. 2020 Dec 14;0(0). Available from: https://www.ijidonline.com/article/S1201-9712(20)32557-1/abstract
- Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nature Microbiology. 2020 Nov;5(11):1403–7. Available from https://www.nature.com/articles/s41564-020-0770-5 See also https://cov-lineages.org/
- Nextstrain | Clade Naming & Definitions [Internet]. Available from: https://docs.nextstrain.org/en/latest/tutorials/SARS-CoV-2/steps/naming_clades.html
- Callaway E. ‘A bloody mess’: Confusion reigns over naming of new COVID variants. Nature. 2021 Jan 15;589(7842):339–339.Available from: https://www.nature.com/articles/d41586-021-00097-w
- Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. 2020 Jul 1;98(7):495–504. Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7375210/
- Plante JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2020 Oct 26;1–6. Available from https://www.sciencedirect.com/science/article/pii/S0092867420308205
- Hodcroft EB, Zuber M, Nadeau S, Crawford KHD, Bloom JD, Veesler D, et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv [Internet]. 2020 Nov 27; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7709189/
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 Dec 22;2020.12.21.20248640. Available from: https://www.medrxiv.org/content/10.1101/2020.12.21.20248640v1
- Faria N, Claro I, Candido D et al .Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings [Internet]. Virological. 2021. Available from: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586
- Resende P, Bezerra J, et al. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020 [Internet]. Virological. 2021. Available from: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584
- Nextstrain | Updated Nextstain SARS-CoV-2 clade naming strategy [Internet]. Virological. 2021. Available from: https://virological.org/t/updated-nextstain-sars-cov-2-clade-naming-strategy/581
- Rambaut A, Loman N, Pybus OG. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations - SARS-CoV-2 coronavirus / nCoV-2019 Genomic Epidemiology [Internet]. Virological. 2020. Available from: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- NCBI Gene Database | ORF1ab ORF1a polyprotein;ORF1ab polyprotein [Severe acute respiratory syndrome coronavirus 2] - Gene - NCBI [Internet]. Available from: https://www.ncbi.nlm.nih.gov/gene/43740578
- Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020 Apr 16;181(2):281-292.e6. Available from:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7102599
- Zinzula L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem Biophys Res Commun [Internet]. 2020 Oct 21; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7577707/
- Li J-Y, Liao C-H, Wang Q, Tan Y-J, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020 Sep;286:198074. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7309931/
- Rambaut A, Loman N. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations [Internet]. Virological. 2020. Available from: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563
- Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell. 2020 Sep 3;182(5):1295-1310.e20. Available from: https://www.sciencedirect.com/science/article/pii/S0092867420310035
- Mathavan S, Kumar S. Evaluation of the Effect of D614G, N501Y and S477N Mutation in SARS-CoV-2 through Computational Approach. 2020 Dec 28 ; Available from: https://www.preprints.org/manuscript/202012.0710/v1
- Yi C, Sun X, Ye J et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies [Internet]. Vol. 17, Cellular & molecular immunology. Cell Mol Immunol; 2020. Available from: https://pubmed.ncbi.nlm.nih.gov/32415260/
- Fratev F. The SARS-CoV-2 S1 spike protein mutation N501Y alters the protein interactions with both hACE2 and human derived antibody: A Free energy of perturbation study. bioRxiv. 2020 Dec 26;2020.12.23.424283. Available from: https://www.biorxiv.org/content/10.1101/2020.12.23.424283v1
- Kemp SA, Harvey WT, Datir RP, Collier DA, Ferreira I, Carabelli AM, et al. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv. 2020 Dec 21;2020.12.14.422555.Available from: https://www.biorxiv.org/content/10.1101/2020.12.14.422555v3
- Hoffmann M, Kleine-Weber H, Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol Cell. 2020 May 21;78(4):779-784.e5. Available from: https://pubmed.ncbi.nlm.nih.gov/32362314/
- Oluniyi P. Detection of SARS-CoV-2 P681H Spike Protein Variant in Nigeria - SARS-CoV-2 coronavirus / nCoV-2019 Genomic Epidemiology [Internet]. Virological. 2020. Available from: https://virological.org/t/detection-of-sars-cov-2-p681h-spike-protein-variant-in-nigeria/567
- Rouzine IM, Rozhnova G. Antigenic evolution of viruses in host populations. PLOS Pathogens. 2018 Sep 12;14(9):e1007291. Available from https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1007291
- Kemp SA, Collier DA, Datir RP. 1 Neutralising antibodies drive Spike mediated SARS-CoV-2 evasion. medRxiv: 2020 Dec 19 Available from: https://www.medrxiv.org/content/10.1101/2020.12.05.20241927v2.full.pdf
- Thermo Fisher | TaqPath COVID-19 Multiplex Diagnostic Solution - [Internet]. Available from: www.thermofisher.com/uk/en/home/clinical/clinical-genomics/pathogen-detection-solutions/covid-19-sars-cov-2/multiplex.html
- Public Health England | Investigation of novel SARS-COV-2 variant Variant of Concern 202012/01 - Technical Briefing 1 [Internet]. 2020. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/947048/Technical_Briefing_VOC_SH_NJL2_SH2.pdf
- Public Health England | Investigation of novel SARS-COV-2 variant Variant of Concern 202012/01 - Technical Briefing 2 [Internet] ; 2020. Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/948152/Technical_Briefing_VOC202012-2_Briefing_2_FINAL.pdf
- Thermo Fisher | FAQ: Impact of the 69-70del mutation in the spike protein of SARS-CoV-2 on TaqPath COVID-19 testing assays [Internet]. Applied Biosystems; Available from: https://assets.thermofisher.com/TFS-Assets/GSD/Reference-Materials/69-70del-s-gene-mutation-ceivd-faq.pdf
- WHO | Covid T, Rt I, Number E. WHO Emergency Use Assessment Coronavirus disease (COVID-19) IVDs. 2020;89. – page 60 https://extranet.who.int/pqweb/sites/default/files/documents/201104_EUL_0525-156-00_TaqPathCOVID-19CE-IVD%20RT-PCR_PR_v_2.0.pdf
- Davies NG, Barnard RC, Jarvis CI, Kucharski AJ, Munday J, Pearson CAB, et al. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England [Internet]. Epidemiology; 2020 Dec. Available from: http://medrxiv.org/lookup/doi/10.1101/2020.12.24.20248822
- Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2021 Jan 4;2020.12.30.20249034. Available from https://www.medrxiv.org/content/10.1101/2020.12.30.20249034v2
- Horby P, Huntley C, Davis N. NERVTAG note on B.1.1.7 severity [Internet]. SAGE meeting paper; Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/955239/NERVTAG_paper_on_variant_of_concern__VOC__B.1.1.7.pdf
- Iacobucci G. Covid-19: New UK variant may be linked to increased death rate, early data indicate. BMJ. 2021 Jan 26;372:n230. Available from: https://www.bmj.com/content/372/bmj.n230
- SARS-CoV-2 lineages - B.1.1.7 report 2021-01-19 [Internet]. 2021. Available from: https://cov-lineages.org/global_report_B.1.1.7.html
- Washington NL, White S, Barrett KMS, Cirulli ET, Bolze A, Lu JT. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. medRxiv. 2020 Dec 30;2020.12.24.20248814. Available from: https://www.medrxiv.org/content/10.1101/2020.12.24.20248814v1
- WQAD8 | Colorado reports first US case of COVID-19 variant initially found in UK [Internet]. wqad.com. Available from: https://www.wqad.com/article/news/health/coronavirus/covid-19-variant-found-colorado/73-ac654aef-16f8-4121-99f0-fe009324d7fd
- Tracking SARS-CoV-2 VOC 202012/01 (lineage B.1.1.7) dissemination in Portugal: insights from nationwide RT-PCR Spike gene drop out data - SARS-CoV-2 coronavirus [Internet]. Virological. 2021. Available from: https://virological.org/t/tracking-sars-cov-2-voc-202012-01-lineage-b-1-1-7-dissemination-in-portugal-insights-from-nationwide-rt-pcr-spike-gene-drop-out-data/600
- Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. The Lancet Infectious Diseases. 2020 May 1;20(5):533–4. Available from: https://www.thelancet.com/journals/laninf/article/PIIS1473-3099(20)30120-1/fulltext with real time data available from https://github.com/CSSEGISandData/COVID-19
- Sabino EC, Buss LF, Carvalho MPS, Prete CA, Crispim MAE, Fraiji NA, et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. The Lancet [Internet]. 2021 Jan 27 ;0(0). Available from: https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(21)00183-5/abstract
- COVID-19 Provisional Counts - Weekly Updates by Select Demographic and Geographic Characteristics [Internet]. 2021. Available from: https://www.cdc.gov/nchs/nvss/vsrr/covid_weekly/index.htm
- CDC. COVID-19 and Your Health [Internet]. Centers for Disease Control and Prevention. 2020. Available from: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/evidence-table.html
- Sze S, Pan D, Nevill CR, Gray LJ, Martin CA, Nazareth J, et al. Ethnicity and clinical outcomes in COVID-19: A systematic review and meta-analysis. EClinicalMedicine [Internet]. 2020 Dec 1;29. Available from: https://www.thelancet.com/journals/eclinm/article/PIIS2589-5370(20)30374-6/abstract
- Zeberg H, Pääbo S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature. 2020 Nov;587(7835):610–2. Available from https://www.nature.com/articles/s41586-020-2818-3
- WHO | SARS-CoV-2 mink-associated variant strain – Denmark [Internet]. WHO. World Health Organization; Available from: http://www.who.int/csr/don/03-december-2020-mink-associated-sars-cov2-denmark/en/
- Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. The Lancet. 2021 Jan 9;397(10269):99–111.Available from https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)32661-1/fulltext
- Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. New England Journal of Medicine. 2020 Dec 31;383(27):2603–15. Available from https://www.nejm.org/doi/full/10.1056/NEJMoa2034577
- Resende P, Bezerra J, Vasconcelos R. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020 [Internet]. Virological. 2021. Available from: https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584
- Gagnon A, Miller MS, Hallman SA, Bourbeau R, Herring DA, Earn DJD, et al. Age-Specific Mortality During the 1918 Influenza Pandemic: Unravelling the Mystery of High Young Adult Mortality. PLoS One [Internet]. 2013 Aug 5;8(8). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3734171/
- SARS-CoV-2 lineages - B.1.351 report [Internet]. Available from: https://cov-lineages.org/global_report_B.1.351.html
- SARS-CoV-2 lineages - P.1 report [Internet]. Available from: https://cov-lineages.org/global_report_P.1.html
Comments
Be the first to post a comment